
第66期 / 本文由奥来恩药政策略与临床卓越中心(CCE)团队原创
FDA法规21 CFR 314 H部分(药物)和21 CFR 601 E部分(生物制品)对用于治疗严重或危及生命的疾病的新药与生物制品加速批准(Accelerated Approval,AA)途径列出了规定。要符合加速批准的条件,药物必须基于充分且有良好对照的临床试验,并且依据流行病学、治疗学、病理生理学或其他证据,证明该产品对替代终点(Surrogate Endpoint)有影响,而且该替代终点可以合理地预测临床获益;或者,基于对生存(Survival)或不可逆发病率(Irreversible Morbidity)以外的临床终点的影响,能够为患者提供比现有治疗更优的效果。如果替代终点与临床获益之间的关系或观察到的临床获益与最终结果之间存在不确定性,申请人希望获得加速批准,就需要对该药物开展进一步研究,以确证其临床获益。上市后研究(Post-marketing Study)通常是需要进行的,并且进行此类研究必须符合“充分且有良好对照”的要求。
如果确证性试验(Confirmatory Trial)取得成功并证明了临床获益,FDA将完全批准(Full Approval)该药物。反之,如果确证性试验未能验证药物的临床获益,申请人需要与FDA的讨论后续计划,可能导致产品被撤出市场。
2023年3月,FDA发布了题为《支持抗肿瘤药物加速批准的临床试验考量》的指南草案。

该指南鼓励申请人修改两项关键性临床试验(Pivotal Study)的设计。针对难治性患者进行的单臂研究,要求用缓解率来支持加速批准,例如总缓解率(Overall Response Rate,ORR)为主要终点(Primary Endpoint),缓解持续时间(Duration of Response,DoR)作为支持终点。在较早阶段进行III期确证性随机对照试验(Randomized Controlled Trial,RCT),以无进展生存期(Progression-Free Survival,PFS)或总生存期(Overall Survival,OS)作为主要终点,验证临床获益并支持完全批准。
由于单臂研究存在严重的局限性,FDA建议在加速批准和完全批准中都使用RCT。FDA建议采用以下两种方法:
(1) 开展两项RCT
-
一项通过早期终点(如ORR)支持药物加速批准。
-
一项则以临床获益终点(如PFS或OS)支持完全批准。
(2) 开展一项RCT
-
在单个研究中,首先分析早期终点(即ORR)以支持加速批准。研究方案应预先确定用于早期分析的随机受试者的数量。然后继续收集这些受试者的后期终点数据,例如OS或PFS,以支持完全批准。研究方案还应该预先设定研究终点的事件数量。
-
方案应包括一项计划,用以可靠地控制支持加速批准的终点和确认临床获益的终点的总体假阳性率(I型误差)。
-
为保持试验过程中数据的完整性,在对早期终点进行分析后,应保持后期终点数据的盲法。
FDA可能要求申请人在加速批准前提供OS数据,以排除药物的有害影响。申请人需要制定一项计划来确保盲法。
对于研发抗肿瘤药物的申请人来说,加速批准可以有效缩短药物获批上市的时间。根据我们以往与FDA沟通互动的经验,FDA对支持加速批准的肿瘤学关键性试验是有明确的预期与要求的。
2023年12月1日,JAYPIRCA®(pirtobrutinib)用于治疗慢性淋巴细胞白血病(Chronic Lymphocytic Leukemia,CLL)和小淋巴细胞淋巴瘤(Small Lymphocytic Lymphoma,SLL)的适应症获得了加速批准(详见FDA的肿瘤学/血液恶性肿瘤批准通知)。在一项开放标签、国际单臂多队列试验中,对108名既往接受过至少两种疗法(包括BTK抑制剂和BCL-2抑制剂)治疗的CLL或SLL患者进行了疗效评估。主要疗效结果指标为ORR和DoR,由独立评审委员会根据2018 iwCLL标准进行评估。患者的ORR为72%(95% CI:63,80),中位DoR为12.2个月(95% CI:9.3,14.7)。上述所有缓解均为部分缓解(Partial Response,PR)。
JAYPIRCA®还于2023年1月27日获得了FDA的加速批准,用于治疗至少接受过两种疗法(包括BTK抑制剂)后复发或难治性的套细胞淋巴瘤(Mantle-cell Lymphoma,MCL)。在一项JAYPIRCA单药治疗的开放标签、多中心、单臂试验中,对120名既往接受过BTK抑制剂治疗的MCL患者进行了疗效评估。主要疗效指标为ORR和DoR,由独立评审委员会根据Lugano标准进行评估。患者的ORR为50%(95% CI:41,59),完全缓解(Complete Response,CR)率为13%。估计中位DoR为8.3个月(95% CI:5.7,NE),6个月时估计DoR率为65.3%(95% CI:49.8,77.1)。

2021年5月5日,KEYTRUDA®(pembrolizumab,帕博利珠单抗)与曲妥珠单抗(trastuzumab)、氟嘧啶和含铂化疗联合用于局部晚期或转移性HER2阳性胃癌患者的一线治疗获得FDA加速批准。这项多中心、随机、双盲、安慰剂对照试验共招募了698名既往没有接受过全身性系统治疗的HER2阳性晚期胃癌或胃食管结合部(Gastroesophageal Junction,GEJ)腺癌患者,并对其疗效进行了评估。与接受安慰剂联合曲妥珠单抗和化疗的患者相比,随机接受KEYTRUDA®联合曲妥珠单抗和化疗的患者的ORR出现了统计学意义明显改善(见下表82)。在存在可用疗法的情况下,FDA预计治疗组替代终点的95%置信区间下限将超过对照组的替代终点,即治疗组ORR的95% CI下限为66%,超过对照组的52%(见下表82)。
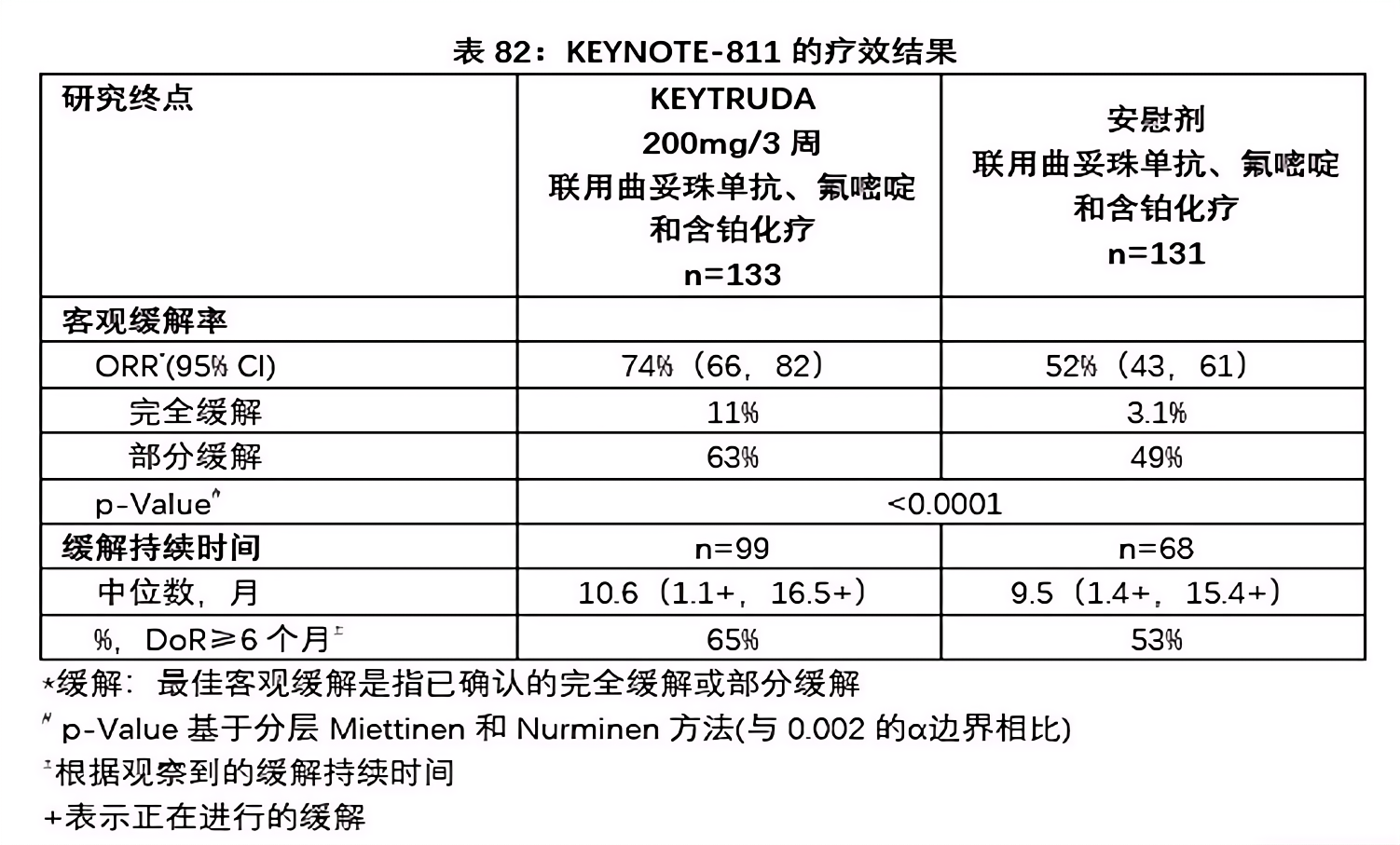
(详细信息请参考Keytruda®说明书)
四、关键性研究的开展速度
FDA在《严重疾病加快上市程序-药品与生物制品(2014年5月)》指南中指出,FDA会在对申请的加快上市程序做出决定时(例如,在NDA或BLA加速批准审评期间)确定现有疗法(Available Therapy)。FDA鼓励申请人在与其互动时讨论关于现有疗法的考量。因此,申请人应快速开展旨在加速批准的关键性研究,并通过 clinicaltrials.gov 和/或竞争对手在科学会议上的发言监测其RCT时间表,争取在现有疗法(即获得全面批准的药物/适应症)发生变化并破坏其加速批准战略之前,获得加速批准。
在研发阶段,申请人与FDA讨论加速批准所需的关键研究时,FDA往往无法明确回答足以支持加速批准的缓解率阈值问题。因为与现有疗法相比,有意义的疗效改善标准是不断变化的。
FDA虽然倾向于将RCT作为加速批准的基础,但仍就单臂研究向申请人提供了一些他们的考量,包括:
-
终点(Endpoints):如果使用的评估终点不是RECIST规定的ORR,申请人应在研究设计阶段与FDA进行讨论。
-
现有疗法(Available Therapy):为了符合加速批准的条件,研究需要在FDA对NDA/BLA审评做决定时证明其与现有疗法相比具有显著优势。申请人应与FDA讨论与现有疗法进行比较的依据。
-
样本量(Sample Size):为了使估计值具有足够的精度,需要提供DoR的可靠估计值,并充分描述药物的不良事件情况。
-
随访时间(Follow-up Time):大多数受试者需要至少6个月的随访时间来估计DoR。
-
预规范(Pre-specification):预先确定样本量和分析人群。
-
疗效评估(Response Assessment):提供盲态独立中心评估(Blinded Independent Central Review,BICR)。
-
缓解率(Response Rate):提供完全缓解和部分缓解的数据(PR+CR)。FDA不接受将疾病稳定(Stable Disease,SD)纳入临床获益率终点。
获得加速批准的药物需要进行上市后确证性试验来验证预期的临床获益。此类试验有助于解决替代终点或中间终点与最终临床获益之间关系的不确定性。为了最大限度地缩短这种不确定性的持续时间,FDA可能会要求申请人在产品获得批准之前,或在批准之后的一段特定时间内,开展旨在验证临床获益的研究。上市后研究必须按照FDA的要求尽职尽责地进行,这些要求可能包括注册目标、研究方案、里程碑和完成研究的目标日期。FDA在数据库(https://www.fda.gov/drugs/resources-information-approved-drugs/ongoing-cancer-accelerated-approvals)中发布了有关上市后要求(Postmarking Requirement,PMR)研究及其计划完成时间表的相关信息。申请人必须在年度报告中提供确证性试验的进展情况。

如果上市后确证性试验未能验证临床获益或未能满足其他条件(例如,其他证据表明该产品在使用时未显示出安全或有效性、申请人未能尽职尽责地进行规定的药品批准后研究等),已获得的加速批准可能会被FDA撤销。如果确证性试验结果为阴性,FDA将会与申请人进行讨论,寻找替代的确证性试验,例如在同一疾病的不同阶段进行研究,并修改PMR。当FDA和申请人无法就替代的确证性试验达成一致时,FDA可要求撤销其加速批准,并在数据库中公布(https://www.fda.gov/drugs/resources-information-approved-drugs/withdrawn-cancer-accelerated-approvals)。

奥来恩拥有经验丰富的药政法规与临床研发专业团队,竭诚为您提供药政法规咨询、注册申报及临床研发服务。我们为您制定获得加速批准的新药研发策略、提供与FDA的有效沟通、高效完成临床研究、以及准备NDA/BLA申请文件与资料。凭借我们团队扎实且丰富的专业知识、为客户定制的解决方案,与您真诚的合作、助力在研新药早日成功上市。欢迎联系我们(邮箱:bd@aleonpharma.com;手机:17761872613)。
(备注:本文中所有提及的药物包括人用药品和生物制品)
